Welcome to Omni-C’s documentation!¶
Overview¶
The Dovetail™ Omni-C™ library uses a sequence-independent endonuclease for chromatin digestion prior to proximity ligation and library generation.
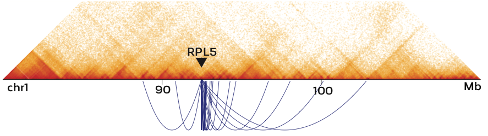
Chromatin interactions: Omni-C Libraries Enable Genome Wide Resolution of Chromatin Interactions. By employing a sequence-independent endonuclease for chromatin digestion, Omni-C™ offers all the characteristics of Hi-C libraries, without the sequence bias inherent to restriction enzyme (RE) based Hi-C approaches. Omni-C™ data contains a significant overlap with data generated using RE based approaches, but are enriched in long-range cis reads. Improved resolution for chromatin conformation and looping interactions can be measured due to this lack of sequence bias. Omni-C™ libraries enable the most complete view of genome-wide chromatin conformation by dramatically increasing resolution of topological interactions that occur in regions with low restriction enzyme density.
Key benefits of Omni-C:
Sequence independent chromatin fragmentation enables fully genome-wide detection of chromatin contacts (up to 20% of the genome lacks coverage using restriction enzyme based Hi-C approaches)
Shotgun sequencing-like even genome coverage enabling SNP calling, chromosome phasing and structural variant detection
Lower sequencing burden to reach desired sequence depth saving time and cost
SNPs and chromosome phasing: The even sequence coverage from Omni-C™ libraries enables genome-wide SNP calling and downstream applications reliant on SNP information, such as chromosome phasing due to low switch error rates. Omni-C™ technology offers the best possible approach for whole genome physical phasing using Illumina short reads.
Large SVs are aaptured in Omni-C™ data: The proximity ligation data can be used to detect and confirm chromosomal rearrangements in cancer samples at a high resolution. Using open-source software tools such as HiGlass, contact matrices enable the quick visualization of such large structural variants.
This guide will take you step by step on how to QC your Omni-C library, how to interparate the QC results and how to generate contact maps, study chromatin structure, use Omni-C data analyzing and enhancing your assembly and more. If you don’t yet have a sequenced Omni-C library and you want to get familiar with the data, you can download Omni-C sequenced libraries from our publicaly available data sets.
The QC process starts with aligning the reads to a reference genome then retaining high quality mapped reads. From there the mapped data will be used to generating a pairs file with pairtools, which categorizes pairs by read type and insert distance, this step both flags and removes PCR duplicates. Once pairs are categorized, counts of each class are summed and reported.
If this is your first time following this tutorial, please check the Before you begin page first.
 The full process including installation, aligning and filtering, library QC, generating contact map and identifying chromatin structures can be completed in less than 48hr compute time for a 800M reads human data set on an Ubuntu 18.04 machine with a 2T volume, 16 CPUs and 64GiB.
The full process including installation, aligning and filtering, library QC, generating contact map and identifying chromatin structures can be completed in less than 48hr compute time for a 800M reads human data set on an Ubuntu 18.04 machine with a 2T volume, 16 CPUs and 64GiB.
Contents: